|
|

|

|
| e-Pub |
Section: New Results
Structural variants
D. Iakovishina defended in 2015 a PhD thesis co-advised by M. Régnier and V. Boeva (Curie Institute). She proposed a new computational method to detect structural variants using whole genome sequencing data. It combines two techniques that are based either on the detection of paired-end mapping abnormalities or on the detection of the depth of coverage. SV-Bay relies on a probabilistic Bayesian approach and includes a modelization of possible sequencing errors, read mappability profile along the genome and changes in the GC-content. Keeping only somatic SVs is an additional option when matched normal control data are provided. SV-Bay compares favorably with existing tools on simulated and experimental data sets [12] Software SV-Bay is freely available https://github.com/InstitutCurie/SV-Bay .
As a side product, a novel exhaustive catalogue of SV types -to date the most comprehensive SV classification- was built. On the grounds of previous publications and experimental data, seven new SV types, ignored by the existing SV calling algorithms, were exhibited.
Structural variations can also be observed and analyzed at larger time scales, and computational methods can be used to predict the structure of ancestral genomes. Within two collaborations with C. Chauve, A. Rajaraman (Simon Fraser University, Canada) and J. Zanetti (SFU, Canada & UniCAMP, Brazil), we revisited the problem of predicting a parsimonious set of adjacencies between ancestral genes, i.e. the most likely structure of an ancestral genome. More specifically, we modified the dynamic programming scheme underlying the DeCo algorithm [28] to compute indicators of robustness for predicting adjacencies. Our reimplementation, which relies on interesting meta-programming strategies, is available at https://github.com/yannponty/DeClone .
In a first study, we postulated a Boltzmann-Gibbs distribution over the set of evolutionary scenarii [9] . Our initial experiments relied on Boltzmann sampling to estimate the probabilities of ancestral adjacencies, but our extended version describes an exact polynomial-time computation of such probabilities, through an adaptation of the inside-outside algorithm. We interpreted such probabilities as supports for predicted adjacencies, and found that discarding adjacencies associated with low supports provided a good strategy for resolving synthenic conflicts.
|
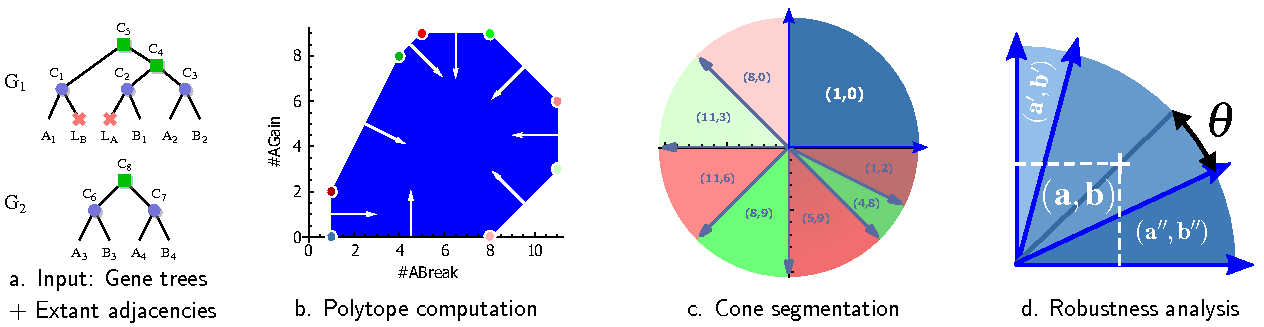
However, the costs associated with the main operations (gaining/breaking adjacencies) in the underlying evolutionary models must be set beforehand in a somewhat arbitrary fashion. This has led us to investigate the influence of those costs on the characteristics of parsimonious predictions, i.e. the robustness of predictions with respect to perturbations of the scoring scheme [18] . To that purpose, we have performed an exact parametric analysis of the DeCo dynamic programming scheme (see Fig. 4 for details). This analysis revealed a quasi-independence, for a large subset of gene trees, of predicted adjacencies to the actual numerical values involved in the scoring scheme.